Programs
IgA Nephropathy
Immunoglobulin A nephropathy (IgAN) is the leading cause of primary glomerulonephritis worldwide, with an estimated incidence of 1.3 per 100,000 individuals per year in the United States.
The global incidence of IgAN is approximately 2.5 per 100,000 individuals per year. Variations in disease incidence and prevalence are, in part, due to regional differences in urine screening, referral patterns and indications for biopsy. We estimate that IgAN is associated with progressive loss of kidney function leading to end-stage kidney disease (ESKD) in approximately 30% to 45% of IgAN patients over 20 to 25 years, representing a significant unmet need for new treatment options.
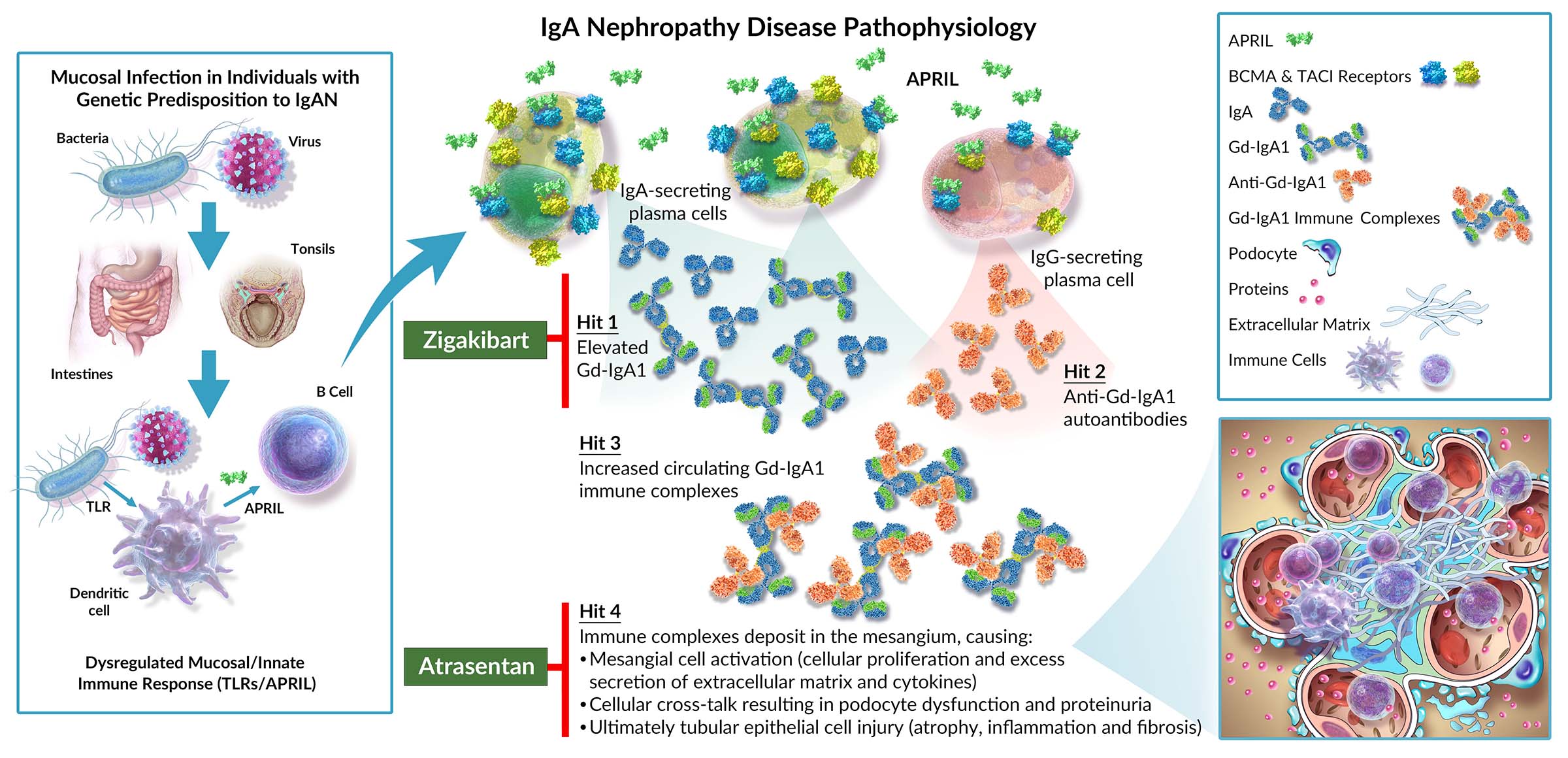
Galactose-deficient immunoglobulin A1, or Gd-IgA1, is recognized as a critical autoantigen to which IgAN patients develop circulating autoantibodies, resulting in the formation and deposition of immune complexes in the glomeruli of the kidney. This process initiates an inflammatory cascade that damages the glomeruli, resulting in protein and blood leaking into the urine, called proteinuria or hematuria, respectively.
Ultimately the filtration function of the kidney is impaired, reducing the ability to remove waste products from the blood. As the disease progresses, these waste products accumulate and can result in potentially life-threatening complications that often lead to the need for dialysis or kidney transplant. Sustained proteinuria is the most widely studied and the strongest predictor for the rate of progression to ESKD in IgAN.